小児難病Ritscher-Schinzel症候群の新たな原因遺伝子と発症メカニズムを解明
名古屋市立大学は7月7日、これまで原因が不明であった小児難病「Ritscher-Schinzel(リッチャー・シンゼル)症候群」において、新たな原因遺伝子を複数特定し、その発症メカニズムを明らかにしたと発表しました。
Ritscher-Schinzel症候群は、頭蓋顔面の特徴、小脳欠損と心血管系の奇形などを特徴とする疾患です。希少な疾患であるため、病歴や分子病態に関する情報が乏しく、たとえ原因遺伝子が特定されても、患者さんの日常診療の改善に繋がりにくいという課題がありました。研究グループは2020年に、この症候群の新たな原因遺伝子としてVPS35Lを同定していましたが、当時は疾患の自然歴や病態メカニズムについては不明な点が多くありました。
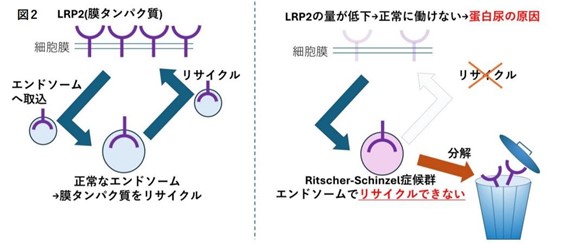
今回、研究グループは、Ritscher-Schinzel症候群の患者さんのゲノムを解析し、COMMD4、COMMD9、CCDC93の3つの遺伝子が、この症候群の新たな原因遺伝子であることを明らかにしました。これらの遺伝子に変異を持つ患者さんには、成長発達の遅れ、脂質異常症、蛋白尿、脳の形成異常など、多岐にわたる合併症が見られることが確認されました。
研究グループは、これらの遺伝子がいずれも、細胞内にある「エンドソーム」という小器官での膜タンパク質のリサイクル機能に関わっていることに注目しました。エンドソームは、細胞が外部から物質を取り込んだり、細胞膜のタンパク質を細胞膜に再び戻したりすることで、細胞の正常な状態を維持する重要な役割を担っています。そこで、ゲノム編集技術を用いてRitscher-Schinzel症候群のモデル細胞やモデル動物を構築し解析した結果、NPxYモチーフを有するLDL受容体やLRP2という膜タンパク質の発現量が低下していることが判明しました。これらの発現低下は、脂質異常症や蛋白尿といった症状の発症に関与していることが示唆されています。さらに、骨や神経組織の発生に不可欠な複数の膜タンパク質の発現低下も確認されています。
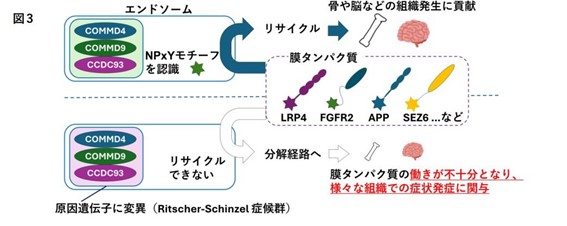
この一連の異常により、膜タンパク質が本来の機能を果たせなくなり、Ritscher-Schinzel症候群に特徴的な多様な合併症を引き起こしていると考えられます。これらの知見に基づき、研究グループは、エンドソームの機能不全を「リサイクル異常症」という新たな疾患概念として提唱しています。
以上の研究成果より、これまで診断がついていなかった患者さんたちが新たにRitscher-Schinzel症候群と診断され、疾患の症状に対する理解を深めることで、より適切な医療やケアの提供に貢献すると考えられます。また、病気の分子メカニズムが明確になったことは、将来的な治療法開発へと繋がることが期待されます。
なお、同研究の成果は、「Science Translational Medicine」に7月3日付で公開されました。