ぎってるまんしょうこうぐんギッテルマン(Gitelman)症候群Gitelman Bartter syndrome
小児慢性疾患分類
- 疾患群2
- 慢性腎疾患
- 大分類13
- ギッテルマン(Gitelman)症候群
- 細分類33
- ギッテルマン(Gitelman)症候群
病気・治療解説
概念
Bartter症候群(BS),Gitelman症候群(GS)は低カリウム血症,代謝性アルカローシスなどを特徴とする先天性尿細管機能障害に伴う症候群である。BSは臨床的には,新生児期に発症する重症型の新生児型と,乳幼児期に発見される比較的軽症型の古典型に分類される。BSは一般的に正常血清マグネシウム値および高カルシウム尿症を呈する。一方,GSでは低マグネシウム血症,低カルシウム尿症を伴い,臨床症状も軽いことでBSと鑑別診断できる。
近年の分子生物学の進歩に伴い,1型~4b型(5型)BSおよびGSに分類されることが示された1~8)。
その結果,従来の臨床分類による新生児型BS,古典型BS,GSという分類がその原因遺伝子変異に伴う臨床症状と必ずしも1対1で対応しないことが明らかとなり,臨床においてその診断に大きな混乱をきたしている。また,二次的要因や他の先天性尿細管異常症に伴う偽性BS,偽性GSの発症頻度は意外に高く,これらの症例における臨床診断は非常に難しい場合がある。
近年はBS,GSを一つの疾患概念と捉え,遺伝性塩類喪失性尿細管機能異常症(salt-losing tubulopathy)と総称する傾向にある4)。
病因
Bartter症候群の原因として,Henle係蹄の太い上行脚でのNaClの輸送に携わるNKCC2,ROMK1,ClC-Kbの三つの遺伝子異常が知られており,それぞれ1,2,3型と分類されている5~8)。
最近,Clチャネル機能発現に必要とされるbarttinの遺伝子(BSND)異常による4型が報告され,この場合,難聴を伴い発症が胎生期と早くかつ重症である2)。その後Schlingmannらにより,ClC-KaとClC-Kbの両方の遺伝子変異を同時に有する場合,4b型(5型)BSとして分類されつつある3)
Gitelman症候群は遠位尿細管上皮細胞膜に発現するNCCTをコードする遺伝子の変異で発症することが示された8)。
症状
本症の症状として,
①乳幼児期発症(常染色体劣性遺伝),②高レニン,高アルドステロン血症による低K血症代謝性アルカローシス③傍糸球体装置過形成,④血圧正常,⑤浮腫がない,⑥アンジオテンシンIIによる昇圧反応の低下,⑦尿中プロスタグランジン排泄増加,⑧尿濃縮障害,⑨成長障害などがみられる。
発症は乳幼児期(生後数年)で,成長障害がみられることも多い。低K血症に起因する尿の濃縮障害も特徴的である。3型は古典的BSともいわれ,胎児期より症状を呈する1,2,4型による新生児型BSに比べると軽症である。
) 1型BS
フロセミドの作用点であるNa+-K+-2Cl-共輸送体NKCC2をコードする遺伝子SLC12A1の異常により発症する。1型は新生児型で,ほとんどの症例で出生前より羊水過多を指摘され,早産,低出生体重で出生する。その後成長障害を伴いやすく,多飲多尿,発熱,嘔吐,脱水などの症状を認める。また,腎性高カルシウム尿症,腎石灰化を認める。末期腎不全へ進行する症例もある。
) 2型BS
カリウム(K)チャネルであるROMKをコードする遺伝子KCNJ1の異常により発症する。羊水過多・早産低出生体重を認め,1型同様に新生児型に分類される。しかし2型においては出生後しばらく高カリウム血症を認めることが知られており,生後3日目前後にピークとなり生後7日目前後には正常値になると報告されている。出生後,高カリウム血症および代謝性アシドーシス,多尿を伴うため,出生直後に2型BSと診断することは非常に困難である。生後数か月より血清カリウム値は低下しはじめるが,1型と比較しカリウムの値低下は比較的穏やかで,正常下限で推移する場合もある。全例において腎性高カルシウム尿症,腎の石灰化を認める。
) 3型BS
クロライドチャネルであるClC-Kbをコードする遺伝子CLCNKBの異常で発症する。3型は一般的に古典型に分類される。乳幼児期に多飲多尿や成長障害で発見され,腎石灰化を認めず,末期腎不全へと進行することはまれとされてきたが,遺伝子診断で3型と診断された患者においても羊水過多,腎石灰化を認め,新生児型を呈する症例が数多く報告されている。
また,尿中カルシウム(Ca)排泄量もほとんどの症例で正常から軽度上昇程度で,低カルシウム尿症を認める症例や,低マグネシウム血症の症例も報告され,このような症例においてはGSとの鑑別診断が非常に難しい場合がある。
) 4型BS,4b型(5型)BS
新生児型を呈し,感音難聴を伴う。典型例においてはBSのなかで最重症型を呈する。BSに感音難聴を伴う群があることは以前から知られており,それらの群では,羊水過多,低出生体重,著明な多尿成長障害,運動発達障害を伴い,非ステロイド性抗炎症薬(NSAID)への反応性が乏しく,幼少時から腎機能障害を認め,末期腎不全へと進行する重症型の臨床経過を辿ると報告されてきた9)。その後,その責任遺伝子および責任蛋白であるBSNDおよびbarttinがクローニングされた。さらにbarttinは腎および内耳に発現していることも示され,腎尿細管においてClc-KaおよびClc-Kbの共通のβサブユニットとして働いていることが判明した1,2)。
最近,CLCNKA,CLCNKBの両方の遺伝子に同時に変異をもつことで,4型BSと同様の臨床像を示すことが報告されており3, 10),これらの症例はbarttinの役割を裏づける結果であり4b型または5型BSと分類される4)。
) GS
Na+-Cl-輸送体NCCTをコードする遺伝子SLC12A3の異常により発症する8)。典型例においてはBSに比較し明らかに軽症である。学童期以降,または成人後に多飲多尿全身倦怠感低カリウム血症や低マグネシウム血症に伴う手足のしびれで発見されることもあるが,偶然の採血で診断される例も多数存在する。また乳幼児期に感冒時の脱水症状で発見される症例や,低身長により診断される症例も存在する。GSは低カルシウム尿症低マグネシウム血症を伴うことからBSと鑑別することができると考えられ,最近までこれらの検査値の違いにより二つの疾患が鑑別されてきた。しかし,前述のように一部の3型BSとGSは臨床像,検査データのみでは鑑別が難しく,また,近年,SLC12A3の遺伝子異常を認めるにもかかわらず,低カルシウム尿症低マグネシウム血症を伴わない症例もあると報告されている。以上のことから,遺伝子診断を行うことではじめて確定診断に至る症例も多数存在する。
診断
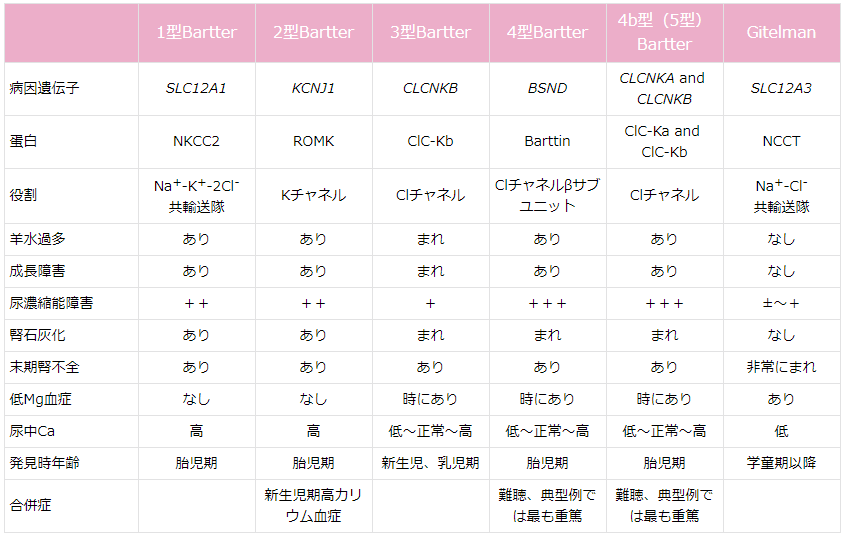
Bartter症候群の原因としては,NaClの輸送にBS,GSは分子生物学的に表1のように分類される。このような臨床的特徴をもとに臨床的および分子生物学的診断を行う。
) 臨床診断
①低カリウム血症,②代謝性アルカローシス,③ 高レニン・高アルドステロン症の三つの条件を満たした際にはじめて本病態を考慮する。そのうえで,出生歴,腎石灰化の有無,血清マグネシウム値,尿中カルシウム値などを参考に病型診断を行う(表1)1-8)。BS,GSともに常染色体劣性疾患であり,両親に同様の病態を認める場合は他の疾患である可能性が高い。BS,GSの全病型において,低身長を伴いやすく,また感冒時の脱水症状,テタニー症状を伴い入院加療を要することが多い。その際の血液検査で診断される症例も多数存在する。
表1 Bartter症候群, Gitelman症候群の病態
) 利尿薬負荷試験
1型BSはフロセミドの作用点であるNKCC2の障害であり,GSはサイアザイドの作用点であるNCCTの障害で発症する。そのため1型BSではフロセミドに無反応,GSではサイアザイドに無反応である。この原理を利用して,利尿薬負荷試験により診断を行うことが可能である。しかし,2型BSにおいては両方の薬剤に反応し,3型BSにおいては従来の予想に反し,フロセミドには反応しサイアザイドに無反応であることが報告され,前述のように,これらの疾患群において最も鑑別の難しい3型BSとGSの鑑別には同検査法は適さないことが判明した11)。
) 遺伝子診断
現在までのところ,遺伝子診断が確定診断を行ううえで最も重要と考えられるが,その変異検出率に関しては不明な点が多い。
) 偽性BS,偽性GS
下剤の長期の使用や,利尿薬の乱用,慢性の咽吐,神経性食思不振症,アルコール中毒などで本病態を呈する。その場合,原因の除去を行っても低カリウム血症の改善には時間を要することが多いため,その鑑別には注意が必要である。
また,腎低形成,ネフロン癆,Dent病,ミトコンドリア病,常染色体優性低カルシウム血症(autosomal dominant hypocalcemia:ADH)などの先天性腎尿細管疾患や嚢胞性線維症,先天性クロール性下痢症において,同様の病態を呈することがあり,その場合BS,GSとの鑑別は非常に困難である。
治療・予後
最も重要なのは低K血症に対する治療である。 K補給薬 (80~300mEq/日)を投与するが,血清Kの上昇はアルドステロン分泌を促すのでアルドステロン拮抗薬であるスピロノラクトン(50~500mg/日)を併用する。アスパラギンを含有するK補給薬は,アスパラギン酸が代謝されてHCO3-となりアルカローシスを助長するので,使用しない。
スピロノラクトン以外にもACE阻害薬,ARBの投与でRAA系を抑制するのはよい。難治例ではプロスタグランジン合成阻害薬も使用されるが,腎機能の悪化を来す恐れがあるため慎重に用いる。
) 新生児型BS
新生児期や乳児期は脱水を伴いやすく,その補正および電解質の補正が必要である。一方,2型BSにおいては新生児期に高カリウム血症を認めるためグルコース・インスリン療法などが必要となる場合がある。低カリウム血症に対しては血清カリウム値の適正化が必要であり,そのためにカリウム製剤(1~10mEq/kg/日),スピロノラクトン(1~1.5mg/kg/日),ACE阻害薬などが有用である。診断が確定し,多尿や低カリウム血症のコントロールに難渋する際はNSAIDによる治療を開始する(インドメタシン0.5~25mg/kg/日)。しかし,その際は腸穿孔などの合併症の報告もあり注意が必要である。
幼児期以降においても,感冒時に容易に脱水や低カリウム血症の悪化をきたし,入院加療が必要になることがしばしばある。また,成長障害,低身長を伴うことが多く,電解質の補正のみで改善しない場合はGH分泌不全を呈していることがあり,GH補充療法が必要な場合がある。また腎不全へと進行することがあり,NSAIDの副作用も考えられるが,NSAIDの投与歴がない症例においても腎不全発症の報告があり,因果関係は不明である。
) 難聴を伴う新生児型BS
典型例においては最も重症の臨床像を呈し,新生児期から重度の脱水を伴う。新生児期および乳児期は大量の輸液および電解質の補正が必要となり,NSAIDの効果はほとんどの場合不十分である。脱水の補正が難しく重度の成長障害を伴う重症例では,救命のために両側腎臓摘出を行い,腹膜透析加療を行うべきとの意見もある。腎機能障害の進行も早く,予後は決して良好とはいえない。
) 古典的BS
治療の基本は血清カリウム値の補正を行うことである。カリウム製剤,スピロノラクトン,ACE阻害薬などが有用である。多尿や低カリウム血症のコントロールに難渋する際は新生児型BS同様にNSAIDによる治療を開始する。NSAIDによる治療はほとんどの場合非常に有効であり,電解質のコントロール,多尿のコントロールが容易となり,成長障害の改善も認める。低身長を伴う症例で,電解質の補正のみでは改善しない場合はGH分泌不全を呈していることがあり,GHによる治療が必要なことがある。
) GS
血清マグネシウム値の低下がある場合,はその補正を行う。マグネシウム製剤としては塩化マグネシウム,硫酸マグネシウム,アスパラギン酸マグネシウムの吸収が優れており推奨されている。わが国で発売されている製剤としては,硫酸マグネシウムやアスパラギン酸マグネシウムとアスパラギン酸カリウムの配合剤が使用される。水酸化マグネシウムの配合剤も有用であるが,アルミニウムを含有するため長期の使用には注意が必要である。酸化マグネシウム製剤も使用されるが,本来潟下剤として使用される薬剤であり,下痢により低カリウム血症のコントロールが難しくなる場合があり注意が必要である。また血清カリウム値の補正も行う必要があるが,その方法は古典型BSと同様である。GSにはQT延長症候群の合併がみられる場合があり,さらなるQT延長の副作用のある薬剤の投与に際しては注意が必要である。腎予後は良好である。
参考文献
1) Birkenhäger R, Otto E, Schürmann MJ, et al. Mutation of BSND causes Bartter syndrome with sensorineural deafness and kidney failure. Nat Genet 29:310-4, 2001
2) Estévez R, Boettger T, Stein V, et al. Barttin is a Cl- channel beta-subunit crucial for renal Cl- reabsorption and inner ear K+ secretion. Nature414:558-61, 2001
3) Schlingmann KP, Konrad M, Jeck N,et al. Salt wasting and deafness resulting from mutations in two chloride channels. N Engl J Med 350:1314-19, 2004
4) Seyberth HW. An improved terminology and classification of Bartter-like syndromes. Nat Clin Pract Nephrol 4:560-7, 2008
5) Simon DB, Bindra RS, Mansfield TA, et al. Mutations in the chloride channel gene, CLCNKB, cause Bartter’s syndrome type III. Nat Genet. 1997 Oct;17(2):171-8. et al.: Nat Genetl7:171-178, l997
6) Simon DB, Karet FE, Rodriguez-Soriano J, et al. Genetic heterogeneity of Bartter’s syndrome revealed by mutations in the K+ channel, ROMK. Nat Genet4:152-6, 1996
7) Simon DB, Karet FE, Hamdan JM,et al. Bartter’s syndrome, hypokalaemic alkalosis with hypercalciuria, is caused by mutations in the Na-K-2Cl cotransporter NKCC2.Nat Genet 13:183-8, 1996
8) Simon DB, Nelson-Williams C, Bia MJ, et al. Gitelman’s variant of Bartter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter.Nat Genet 12:24-30, 1996
9) Jeck N, Reinalter SC, Henne T, et al. Hypokalemic salt-losing tubulopathy with chronic renal failure and sensorineural deafness. Pediatrics 108: E5, 2001
10) Nozu K, Inagaki T, Fu XJ,et al. Molecular analysis of digenic inheritance in Bartter syndrome with sensorineural deafness. J Med Genet 45: 182-186, 2008
11) Nozu K, Iijima K, Kanda K, et al. The pharmacological characteristics of molecular-based inherited salt-losing tubulopathies. J Clin Endocrinol Metab95:E511-8, 2010
12)Colussi G, Bettinelli A, Tedeschi S, De Ferrari ME, Syrén ML, Borsa N, Mattiello C, Casari G, Bianchetti MG. A thiazide test for the diagnosis of renal tubular hypokalemic disorders. Clin J Am Soc Nephrol. 2:45-60,2007
小児慢性特定疾患情報センターhttps://www.shouman.jp/より、許可をいただき掲載しております。
この疾患に関するピックアップ記事、イベントはありません