先天性ミオパチーの発症機構を新たに解明
岡山大学大学院と国立精神・神経医療研究センターの共同研究グループは、先天性ミオパチー (指定難病 111) の一種である中心核ミオパチーについて、筋肉のT管の形成異常が起こる仕組みを見出したと発表しました。本研究結果により、中心核ミオパチーの早期診断や新たな薬の開発に繋がると期待されます。
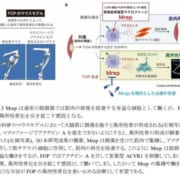
中心核ミオパチーは先天性ミオパチー(指定難病111)の一種で、異常のある遺伝子の種類によって複数の型に分類されます。遺伝子異常のため骨格筋をつくるタンパク質の欠損や活性低下により、筋力が弱く力が入りにくい症状がみられます。呼吸や嚥下の障害、発育遅延を伴うこともあります。中心核ミオパチーは骨格筋を構成するT管に異常がみられます。T管は筋肉を動かすための指令を伝達する役割を持ちますが、ここが障害されることで指令が伝わらず筋肉を動かせなくなります。中心核ミオパチーの原因遺伝子としていくつかの遺伝子が特定されていますが、これらの転写産物の異常によってミオパチーが発症するメカニズムは解明されていません。
本研究ではマウスの筋細胞を用いて、中心核ミオパチーの原因遺伝子として知られるDNM2とBON1がコードするダイナミン2とBIN1によるT管構造形成メカニズムを解析しました。ダイナミン2とBIN1は細胞膜の変形や切断に関与することが知られています。解析の結果、ダイナミン2の発現量を増やすとT管構造が安定化し、ダイナミンの発現量を減少させるとT管の形成が阻害されることが明らかになりました。また、ダイナミン2とBIN1の精製タンパク質を用いて相互作用を調べたところ、BIN1はダイナミン2に結合しGTOアーゼ活性を抑制することでT管構造を安定させていることが示されました。一方で中心核ミオパチー型のダイナミン2はBIN1による制御を受けずに、GTPアーゼ活性は促進されていました。近年徐々に中心核ミオパチー患者の遺伝子変異が多数見つかっています。こうした研究が重ねられることで、先天性の筋疾患に対する新たな診断法や治療薬の開発に繋がると期待されます。
出典元
岡山大学 プレスリリース