せがわびょう瀬川病Segeawa syndrome; Dopamine-responsive dystonia; DRD; Hereditary progressive dystonia with diurnal variation; HPD
小児慢性疾患分類
- 疾患群11
- 神経・筋疾患
- 大分類29
- 変形性筋ジストニー
- 細分類83
- 瀬川病
病気・治療解説
概要
瀬川病は1971年に瀬川ら1)が、「著明な日内変動を呈する遺伝性進行性大脳基底核疾患」として世界ではじめて小児例を報告した。
1990年に藤田・新宅2)により瀬川病患者の髄液中プテリジン濃度の低下が報告され、
GTPシクロシドロラーゼⅠ(GTPCH)活性の低下が指摘されていたが、1994年に一瀬ら3)により
瀬川病患者でGTPCH遺伝子(GCH1)の変異が片方のアリルに発見され、GTPCHの部分欠損によって起こることが明らかにされた。
遺伝性ジストニアとしてDYT5に分類されていたが、病因は補酵素ビオプテリンの合成障害による神経伝達物質病であり、
現在では先天代謝異常症と考えられている。第14染色体(14q22.1-22.2)に存在するGCH1の変異により発症する常染色体優性遺伝性疾患で、
黒質線条体ドパミン神経系終末部のドパミン欠乏による固縮型筋緊張異常によるジストニア姿勢およびジストニア運動を主症状とする。
典型例は10歳以下に発症するが成人発症例も報告されている。女性優位の性差を有する(男:女=1:4)。
ジストニア、とくに小児期の姿勢ジストニアは著明な日内変動を呈する。
病因
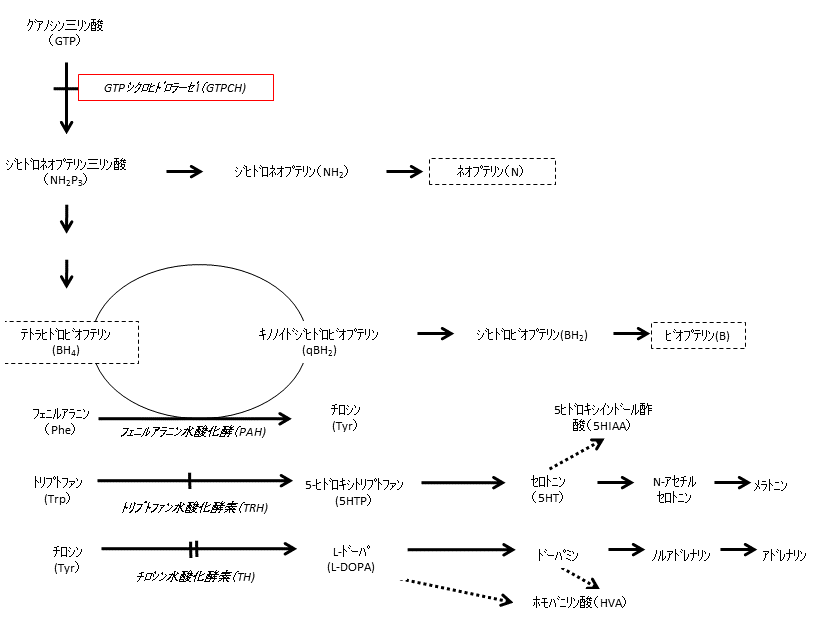
瀬川病の病因は、発達早期に活性化するプテリジン代謝の律速酵素GTPCHの遺伝子変異による活性低下に起因する(図1)。
GTPCHはテトラヒドロビオプテリン(BH4)合成の最初の反応を触媒する酵素で、GTPからジヒドロネオプテリン三リン酸(NH2P3)を産生する。
この酵素蛋白の変異はNH2P3の産生低下から最終産物であるBH4の欠乏をまねき、BH4を補酵素とする芳香族アミノ酸水酸化反応の低下をきたす。
このようにGTPCH欠損症ではGCH1の変異により芳香族アミノ酸水酸化酵素に共通の補酵素BH4の欠乏をきたすが、
優性遺伝形式をとる瀬川病では片方の対立遺伝子は正常(ヘテロ)であるため両方の対立遺伝子に変異(ホモ)のある
劣性遺伝形式をとるBH4欠損症に比べてGTPCH活性の低下は軽度である4)。
このためBH4の低下も軽度で病初期には髄液中のプテリジン値に異常の認められないこともあるが、
症状の進行にともない徐々に低下するようになる。血液や尿ではプテリジン値にあまり変化を認めないが、
症状が進行すると低下が認められるようになる。
BH4は3種の芳香族アミノ酸水酸化酵素の補酵素として作用するが、そのミカエリスメンテン(Michaelis-Menten:Km)値はそれぞれ異なるため、
BH4の低下が軽度であればKm値の一番高いチロシン水酸化酵素だけが障害を受け、カテコールアミンの低下による
ジストニアの症状が発症することになる。瀬川病でもGCH1の変異部位によりBH4産生がより障害される場合にはBH4はさらに低下し、
2番目にKm値の高いトリプトファン水酸化酵素も障害され、セロトニン産生の低下による症状も加わり動作ジストニアが発症する。
しかし最もKm値の低いフェニルアラニン水酸化酵素への影響は少なくいわゆる高フェニルアラニン血症
(血漿フェニルアラニン値>2mg/dL)は発症しない。
図1
疫学
日本で行われた2009年の全国調査で117人の瀬川病患者が報告5)され、世界的な頻度(1/1,000,000)と
ほぼ同じ頻度と考えられていたが、その後毎年20人程度の新しい患者が診断されており、
現在では250程度の患者が診断され日本では50万人に1人の頻度でと考えられる。
そのうち約半数が成人であるが病歴からは小児期にすでに症状の認められていた症例も多い。
臨床症状
発症年齢は10歳未満で男女比は1:4で女児に多く、夕刻に症状が悪化する著明な日内変動があり、
症状は一側の下肢から始まり同側の上肢に広がり対側の下肢そして上肢というようなN字型の進行性のジストニアが特徴である。
姿勢ジストニア型と動作ジストニア型の2型に分けられる6)。
姿勢ジストニア型は、多くは6歳頃、一側下肢内反尖足で発症、15歳頃までに全肢にひろがり、20歳頃まで筋強剛が進行するが、
その後、進行は緩やかになり、30歳以後は定常状態となる。10歳頃から姿勢振戦が認められる。
動作ジストニア型は、姿勢ジストニアに加え、8歳以後、上肢のジストニア運動、頸部後屈、
眼球回転発作(oculogyric crisis:OGC)が発現、思春期以後、主に成人年齢で斜頸、書痙を併発する。
この病型には運動誘発性ジストニア、むずむず足症候群を呈する症例もある。
さらに、成人年齢で斜頸、書痙、または、パーキンソン病様症状で発症する症例がある。
検査所見
一般検査所見及び脳の画像所見では特に異常を認めない。
髄液ホモバニリン酸(homovanilic acid:HVA)は低値(<-2SD)を認めるが、髄液5ヒドロキシ酢酸
(5-hydroxy indole acetic acid:5HIAA)は正常のことが多い。姿勢ジストニアの重症例や動作ジストニアでは低値(<-2SD)を取ることもある。
これらの正常値は年齢により異なるため対象年齢の正常値と比較して評価する必要がある。(年齢別正常値:表1)
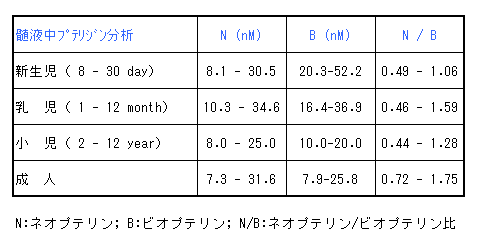
髄液プテリジン分析では、ネオプテリン(N)値とビオプテリン(B)値の両方が低値(<-2SD)を示すがN/B比は正常である。
これらの正常値は年齢により異なるため対象年齢の正常値と比較して評価する必要がある。(年齢別正常値:表2)
遺伝子解析ではGCH1遺伝子の変異が1つのアレルに同定される。
表1:髄液中モノアミン代謝産物
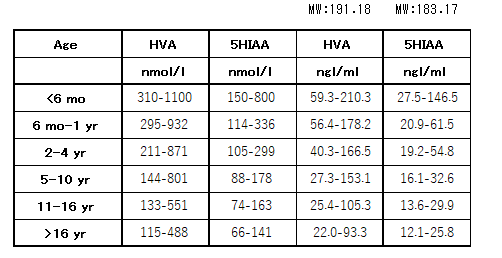
5HIAA = 5-hydroxyindoleacetic acid; HVA = homovanillic acid
1. Hyland K, Biaggioni I, Elpeleg ON, Nyggard TG, Gibson KM. Disorders of neurotransmitter metabolism.
In: Blau N, Duran M, Blaskovics M, eds. Physician’s Guide to the Laboratory Diagnosis of Metabolic Diseases.
London: Chapman & Hall, 1996: 79-98.
2. Blau N, Barnes I, Dhondt JL. International database of tetrahydrobiopterin deficiencies. J Inherit Metab Dis 1996;19:8-14.
3. Blau N, Blaskovics M. Hyperphenylalaninemia. In: Blau N, Duran M, Blaskovics M, eds.
Physician’s Guide to the Laboratory Diagnosis of Metabolic Diseases. London: Chapman & Hall, 1996: 65-78.
表2:髄液中プテリジンの正常値
診断の際の留意点
優性遺伝形式をとり家族内発生が認められるため、病歴については家族歴、特に母親や祖母についても
同様の症状の有無を聴取することが重要である。発症初期の髄液中のHVAやプテリジン分析に異常を認めないことがあり、
数ヶ月から数年の経過で異常値を示すことがあるため、生化学的検査で異常がなくても臨床的に疑われる場合は遺伝子検査を行う。しかし、GCH1遺伝子解析でも変異を認めない症例が10%程度存在する。
治療
姿勢ジストニア型ではL-DOPAが著効を呈し、その効果は副作用なく永続する。末梢のアミノ酸脱炭酸酵素の阻害剤である
カルビドーパの合剤を使用するとL-DOPAとしての投与量は少なくて済む。いずれも保険収載されている。
動作ジストニア型ではL-DOPAに加えて早期から5ヒドロキシトリプトファン(5-hydroxy tryptophan:5-HTP)の投与が望まれる。
5-HTPは薬剤としては認められておらずこれまでは実験用試薬を使用していたが、最近では海外で健康補助食品として販売されているため、
この5-HTPカプセルの使用が価格も安く勧められる。
合併症
動作ジストニア型では家系により鬱病を合併することがある。また、早期、主に乳児期発症例ではセロトニン欠乏を発現、自閉傾向、
うつ傾向、強迫神経症、頭痛を併発する例がある。また、筋緊張低下、ロコモーションの障害をきたし、さらに、脚橋被蓋核活性低下を併発、
ドパミン欠乏により思春期以後パーキンソン病と同様の病状を呈することが知られている。
予後
診断がつけばL-DOPAによる治療が可能であり予後は一般に良好である。
成人期以降の注意点
成人期以降もL-DOPAによる治療が必要で有り、怠薬や治療の中断により症状が再発し悪化することがあるため、生涯を通じて注意深い治療と経過観察が必要である。
参考文献
瀬川昌也:L-DOPAが著効を呈した小児基底核疾患-著明な日内変動を伴った遺伝性進行性基底核疾患-、診療、24:667-672, 1971
藤田繁、新宅治夫:著名な日内変動を伴う遺伝性進行性ジストニア(HPD:瀬川病)の病因とプテリジン代謝、市立釧路医誌、2巻1号頁64ー67, 1990
Ichinose H, Ohye T, Takahashi E, et al.: Hereditary progressive dystonia with marked diurnal fluctuation caused by mutations in the GTP cyclohydrolase I gene. Nature Genetics 8:236-242, 1994
新宅治夫:バイオプテリンと小児神経疾患、脳と発達、41(1): 5 -10, 2009
新宅治夫:小児神経伝達物質病の診断基準の作成と患者数の実態調査に関する研究報告、pp1-83、厚生労働科学研究補助金 難治性疾患克服研究事業 平成21年度総括・分担研究報告書(研究代表者 新宅治夫)、大阪、平成22年3月
瀬川昌也:新生児期発症の神経伝達物疾患. 脳と発達. 43:352-358, 2011
小児慢性特定疾患情報センターhttps://www.shouman.jp/より、許可をいただき掲載しております。
この疾患に関するピックアップ記事、イベントはありません