ぷりおんびょうプリオン病Prion diseases
指定難病23
小児慢性疾患分類
- 疾患群-
- -
- 大分類-
- -
- 細分類-
- -
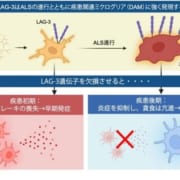
プリオン病
孤発性プリオン病
特発性孤発性クロイツフェルト・ヤコブ病
孤発性クロイツフェルト・ヤコブ病
遺伝性クロイツフェルト・ヤコブ病
遺伝性プリオン病
家族性クロイツフェルト・ヤコブ病
ゲルストマン・ストロイスラー・シャインカー病
致死性家族性不眠症
獲得性プリオン病
クールー
医原性クロイツフェルト・ヤコブ病
変異型クロイツフェルト・ヤコブ病
実施中の治験/臨床試験
募集中
- プリオン病患者を対象にION717の髄腔内投与の安全性、 忍容性、薬物動態及び薬力学を評価する第1/2a相試験|プリオン病|令和8年4月10日公表
(最終更新:2026年5月13日)