むこたとうしょうなながたムコ多糖症Ⅶ型Mucopolysaccharidosis
小児慢性疾患分類
- 疾患群8
- 先天性代謝異常
- 大分類6
- ライソゾーム病
- 細分類80
- ムコ多糖症Ⅶ型
病気・治療解説
概要
ムコ多糖症VII型は、低身長、関節拘縮、心臓弁膜症、精神運動発達遅滞、胎児水腫など多彩な症状を示す。頻度は極めてまれである。ライソゾーム内に存在するβ-グルクロニダーゼ酵素活性が欠損するため、ケラタン硫酸を除くすべてのムコ多糖の分解が障害される常染色体劣性遺伝性疾患である。
疫学
きわめてまれな疾患で現在確認されている日本人生存症例は5例以下である。
病因
ライソゾーム内に存在するβ-グルクロニダーゼ酵素活性が原因で、ケラタン硫酸を除くすべてのムコ多糖の分解が障害され蓄積する進行性全身性疾患である。
症状
ムコ多糖症に特徴的な骨変形、関節拘縮を認める。肝腫大、角膜混濁、知的障害、心臓弁膜症は軽度であり、認められないこともある。尿中ウロン酸は多くの小児例ではやや増加するが著明ではなく、成人ではほとんど増加を認めない。
診断
① 尿中ムコ多糖分析:尿中のグルコサミノグリカン(ウロン酸)の量が増加するが、軽症型や成人では増加は認められない。ヘパラン硫酸、デルマタン硫酸、コンドロイチン硫酸の量が増加するが、総ウロン量としては多くない。
② β-グルクロニダーゼ活性:末梢リンパ球あるいは白血球、培養皮膚繊維芽細胞中のβ-グルクロニダーゼ活性の欠損あるいは低下する。
治療
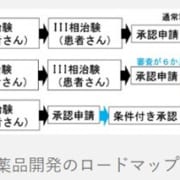
対症療法と原因療法がある。原因療法として造血細胞移植が考慮される場合がある。酵素補充療法は、欧米で開発中である。
予後
進行性で致死性の重篤な疾患である。
成人期以降
進行性疾患のため成人期にはかなり重症化する。造血細胞移植も病態の進行を完全に阻止することはできない。
参考文献
厚生労働省難治性疾患等政策研究事業ライソゾーム病に関する調査研究班編集「ライソゾーム病・ペルオキシソーム病診断の手引き」診断と治療社(2015)
小児慢性特定疾患情報センターhttps://www.shouman.jp/より、許可をいただき掲載しております。
この疾患に関するピックアップ記事、イベントはありません